
Foundation Models and the Power of Proprietary Experimental Data
In recent years, foundation models have revolutionised fields from natural language processing to image generation. Now, these powerful AI systems are transforming drug discovery. By learning generalisable patterns across chemical and biological data, foundation models are unlocking new opportunities to identify novel small molecules and biologics, improve virtual screening accuracy, and accelerate lead optimisation.
However, the true power of foundation models lies not only in their architecture, but in the data used to train them. In this blog, we explore how foundation models are reshaping the pharmaceutical R&D landscape—and why companies with access to high-quality, proprietary experimental data hold a critical advantage.
What Are Foundation Models in Drug Discovery?
Foundation models are large-scale, pre-trained neural networks that learn universal representations from diverse data sources. In drug discovery, these models are trained on small molecule structures, protein sequences, biological assay data, omics profiles, and more. Once trained, they can be fine-tuned for a variety of downstream tasks: target identification, virtual screening, de novo design, and lead optimisation.
These models rely on a variety of architectures:
- Graph Neural Networks (GNNs) represent molecules as graphs and have shown strong performance in structure–activity prediction (Zhu et al., 2023).
- Transformers learn from SMILES strings or protein sequences, capturing chemical and biological syntax.
- Diffusion models are emerging as state-of-the-art tools for generating 3D molecular structures (Hoogeboom et al., 2022).
- Multimodal models integrate different representations (e.g. 2D structures, 3D conformations, and textual annotations) to generate robust molecular embeddings (IBM Research, 2023).
Training strategies include self-supervised learning (e.g. masked token prediction or contrastive learning), multitask learning across ADMET endpoints, and fine-tuning on specific tasks such as property prediction or bioactivity classification (Feinberg et al., 2022).
The Data Behind the Models
While architecture matters, data quality is the key determinant of model performance. Most foundation models are initially trained on publicly available datasets:
- ChEMBL: Curated bioactivity data (~2.4M compounds) (Gaulton et al., 2017)
- PubChem BioAssay: HTS results with millions of activity labels (Wang et al., 2017)
- BindingDB: Binding affinity measurements for drug–target pairs (Liu et al., 2007)
- Protein Data Bank (PDB): 3D structures of proteins and ligand complexes (Burley et al., 2022)
- AlphaFold DB: Predicted structures for over 200M proteins (Varadi et al., 2022)
- LINCS/L1000: Gene expression responses to chemical/genetic perturbations (Subramanian et al., 2017)
Yet these datasets have major limitations:
- Inconsistent assay protocols and missing metadata
- Sparse coverage and lack of true negative data
- Label noise, especially in HTS data
- Bias towards well-studied targets and chemotypes
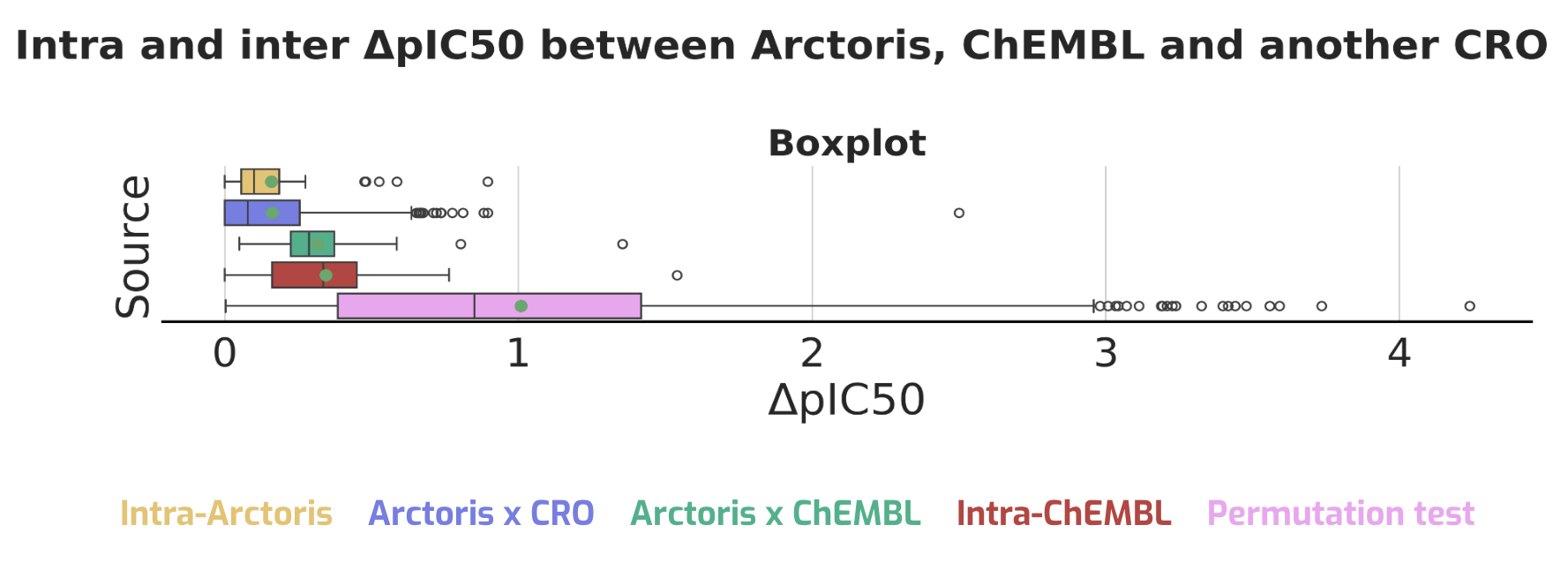
These limitations lead to foundation models that overfit, underperform in generalisation, and struggle with real-world prediction tasks.
The Strategic Value of Proprietary Data
To build truly powerful and generalisable models, companies must go beyond public datasets. Proprietary experimental data—generated through automation and standardisation—offers a clear strategic edge:
- Higher fidelity: Robotic execution ensures consistency and reproducibility across experiments.
- Greater depth: High-resolution data capture (e.g. full dose–response curves, multi-modal readouts) provides richer supervision for training.
- Balanced datasets: In-house screens can be designed to include both actives and inactives, avoiding label imbalance.
- Multi-modal annotations: Linking chemical structure to imaging, transcriptomic, and phenotypic data allows models to learn holistic representations.
- Closed-loop feedback: Tight integration between modelling and experimentation enables active learning, rapidly improving performance in regions of uncertainty.
Conclusion
Foundation models are rapidly becoming central to the future of drug discovery. However, the race will not be won by model architecture alone. The winners will be those with access to clean, high-resolution, and deeply annotated datasets—especially when coupled with the infrastructure to generate more, faster.
As we enter the age of AI-native drug discovery, companies that own their experimental data pipelines and integrate them tightly with model development will hold a formidable advantage.
At Arctoris, we believe that the lab is not just a place to validate predictions—it is the foundation upon which transformative AI is built.
References
Burley, S. K. et al. (2022) RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules. Nucleic Acids Research, 50(D1), pp. D437–D451.
Exscientia (2021) Exscientia announces the first AI-designed drug to enter Phase 1 trials. [Press release]
Exscientia (2023) Drug design and discovery using AI: Case studies and pipelines. [Company blog]
Feinberg, E. N. et al. (2022) Learning Deep Representations of Molecular Conformations. Journal of Chemical Information and Modeling, 62(6), pp. 1173–1186.
Gaulton, A. et al. (2017) The ChEMBL database in 2017. Nucleic Acids Research, 45(D1), pp. D945–D954.
Hoogeboom, E. et al. (2022) Equivariant Diffusion for Molecule Generation in 3D. International Conference on Machine Learning (ICML).
IBM Research (2023) Accelerating drug discovery with multimodal foundation models. [Research blog]
Liu, T. et al. (2007) BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Research, 35(suppl_1), pp. D198–D201.
Mervin, L. et al. (2023) Multitask Learning for Drug Discovery: A Review. Journal of Medicinal Chemistry, 66(5), pp. 3140–3162.
Rao, R. et al. (2021) MSA Transformer. bioRxiv. https://doi.org/10.1101/2021.02.12.430858
Subramanian, A. et al. (2017) A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell, 171(6), pp. 1437–1452.
Varadi, M. et al. (2022) AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research, 50(D1), pp. D439–D444.
Wang, Y. et al. (2017) PubChem BioAssay: 2017 update. Nucleic Acids Research, 45(D1), pp. D955–D963.
Zhang, H. et al. (2021) TxGNN: Representation Learning for Therapeutics. NeurIPS Workshop on Machine Learning in Drug Discovery.
Zhu, H. et al. (2023) MolE: A Graph Transformer Framework for Molecules. Proceedings of ICLR 2023.
Zhavoronkov, A. et al. (2023) AI-driven drug discovery: Discovery of a novel inhibitor of fibrosis. Nature Biotechnology, 41(5), pp. 654–662.